Polycystic kidney disease (PKD) isn’t just a slow decline in kidney function-it’s a genetic time bomb. If you have it, your kidneys grow hundreds, sometimes thousands, of fluid-filled cysts over time. These aren’t harmless bumps. They keep expanding, crushing healthy tissue, and eventually steal your kidneys’ ability to work. About 500,000 people in the U.S. live with PKD, and for many, it ends in dialysis or transplant. But here’s the thing: knowing what’s happening inside your body, and acting early, can change the game.
Two Types, One Root Cause
Not all PKD is the same. There are two main types, and they’re as different as night and day.
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is the big one. It makes up over 98% of all cases. You only need one copy of a broken gene-either PKD1 or PKD2-to get it. If one of your parents has it, you have a 50% chance of inheriting it. Most people don’t feel symptoms until their 30s or 40s, but damage starts decades earlier. The PKD1 mutation tends to be worse. People with it often lose kidney function by age 50-60. Those with PKD2 might hold off until their 70s.
Autosomal Recessive Polycystic Kidney Disease (ARPKD) is rare. You need two broken copies of the PKHD1 gene-one from each parent. Parents usually don’t show symptoms. But if both carry the gene, each child has a 25% chance of being born with ARPKD. This type hits early. Babies can be born with huge kidneys, high blood pressure, and breathing problems. Many don’t survive past infancy. Those who do often face liver scarring and kidney failure by childhood.
What Happens Inside Your Kidneys?
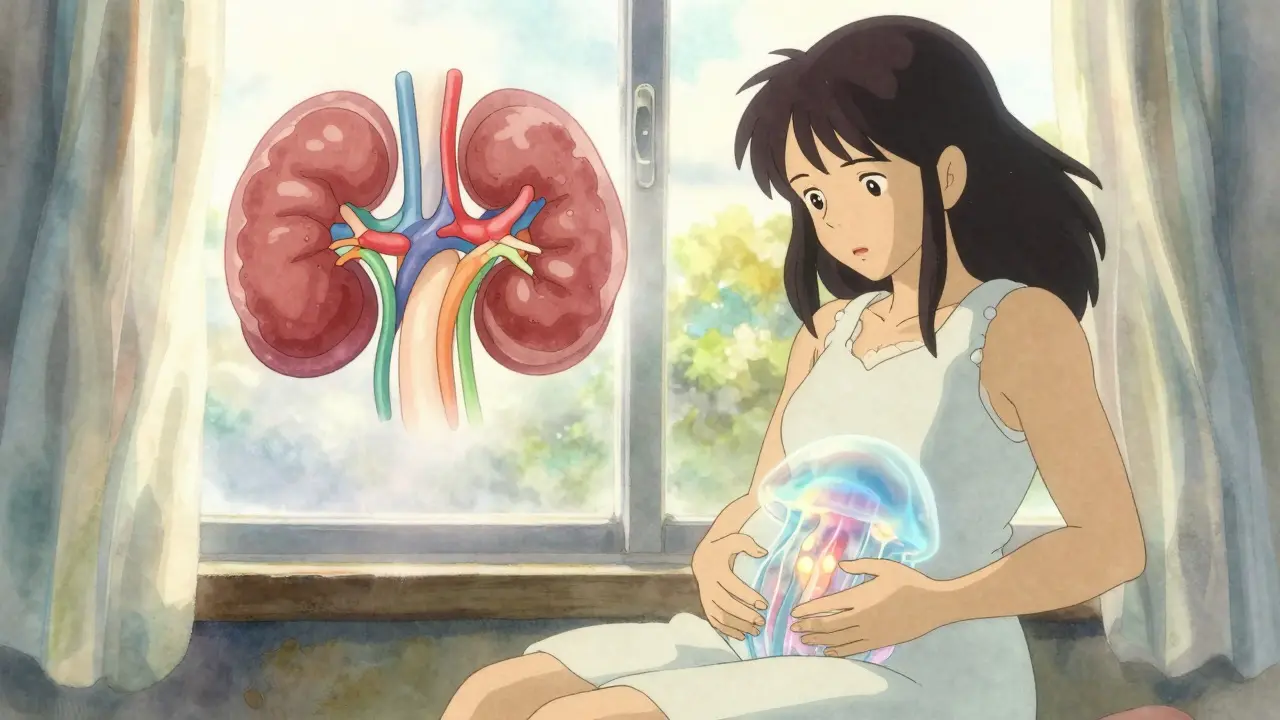
Your kidneys normally filter waste and balance fluids. In PKD, the cells lining the kidney tubules start acting weird. Instead of forming tight tubes, they multiply uncontrollably and form cysts. These cysts grow like balloons, pushing aside healthy tissue. Over time, your kidneys can balloon to three times their normal size-and weigh up to 30 pounds.
It’s not just about size. The cysts leak fluid, trigger inflammation, and mess with blood flow. That’s why high blood pressure shows up early-even before kidney function drops. And because the kidneys can’t filter well, toxins build up. Fatigue, nausea, and swelling follow. By the time people feel really sick, years of damage are already done.
Diagnosis: Finding the Problem Before It’s Too Late
Doctors don’t wait for you to collapse. If you have a family history of PKD, they’ll check you early. The standard tools are simple:
- Ultrasound: The first test. It shows cysts as dark spots. By age 30, someone with ADPKD typically has 10+ cysts per kidney.
- CT or MRI: Used if the ultrasound is unclear or if cysts are small. These show exact size and growth rate.
- Genetic testing: A blood or saliva test checks for mutations in PKD1, PKD2, or PKHD1. It’s not always needed-but if you’re planning a family or have unusual symptoms, it’s powerful.
Here’s a real-world example: A 28-year-old man with no symptoms gets an ultrasound because his dad had PKD. The scan shows 8 cysts. He’s told: "You have ADPKD." That’s not a death sentence. It’s a heads-up. Now he can start managing it.

Managing PKD: What Actually Works
You can’t reverse the cysts. But you can slow them down. And that’s everything.
Control your blood pressure. This is non-negotiable. High pressure crushes the kidneys faster. The target? Below 130/80 mmHg. Some experts now push for even lower-110/75-if you’re young and your kidneys are still working. Medications like ACE inhibitors or ARBs are first-line. They do double duty: lower pressure and protect kidney tissue.
Take tolvaptan (Jynarque). This is the first drug approved to directly slow PKD progression. It blocks a hormone (vasopressin) that tells cysts to grow. Clinical trials showed it cuts kidney function decline by about 1.3 mL/min/year. That might sound small, but over 10 years, it can delay dialysis by 5-10 years. The catch? It costs about $115,000 a year. Insurance often covers it for people with rapid progression-confirmed by kidney imaging and eGFR decline.
Watch your kidneys. Get an eGFR test at least once a year. If it drops below 60 mL/min, test every 3 months. Track it like a stock chart. A steady drop means you’re in the danger zone. A stable number? You’re winning.
Hydrate. Avoid caffeine. Skip NSAIDs. Drink water-lots of it. Caffeine (coffee, soda, energy drinks) can make cysts grow. NSAIDs like ibuprofen? They can spike blood pressure and hurt your kidneys. Use acetaminophen instead.
Don’t smoke. Don’t get obese. Smoking speeds up kidney damage. Obesity raises blood pressure and insulin resistance. Both make PKD worse.
The Human Side: Pain, Anxiety, and Isolation
Most people with PKD don’t die from kidney failure. They die from the weight of waiting for it.
Chronic pain is the #1 complaint. Cysts press on nerves, stretch the kidney capsule, and sometimes bleed or get infected. One patient survey found 78% of people with ADPKD suffer moderate to severe pain. It’s not "just back pain." It’s constant, dull, and deep-like someone’s squeezing your insides.
And then there’s the anxiety. You know your kidneys are failing. You’ve seen your parent on dialysis. You’re terrified your kids will inherit it. A 63% of patients report anxiety about future kidney failure. That’s not normal worry. It’s trauma.
And diagnosis delays? Common. One Reddit user waited 7 years before getting a correct diagnosis-even though his dad had PKD. Doctors dismissed his pain as "stress." That’s not rare.
What’s Next? Hope on the Horizon
There’s no cure yet. But research is moving fast.
Tolvaptan was just the start. New drugs are in phase 3 trials:
- Lixivaptan: Similar to tolvaptan, but potentially safer and cheaper. Results expected in 2024.
- Bardoxolone methyl: Targets inflammation and cellular stress. Early data shows it improves kidney function by nearly 5 mL/min over placebo.
Genetic testing is getting cheaper-around $1,200 now. That means more people can find out if they carry the gene before symptoms start. And for couples planning kids, preimplantation genetic testing can help them have children without PKD.
Even better: the HALT-PKD study proved that aggressive blood pressure control (targeting 110/75) slowed kidney growth by 14.2% over five years. That’s huge. It means lifestyle + medication can buy you decades.
When It’s Time for Transplant or Dialysis
About 50% of ADPKD patients need dialysis or transplant by age 60. For ARPKD, it’s often much earlier-sometimes by age 10.
Dialysis isn’t a cure. It’s a bridge. It keeps you alive, but it’s exhausting. Transplant is better. A new kidney lasts 15-20 years on average. Most PKD patients do well after transplant because the disease doesn’t affect the new organ.
Wait times vary. In the U.S., it’s 3-5 years. Blood type matters. O-negative patients wait longer. Location matters too. Some regions have shorter lists.
But here’s the truth: if you’re managing your PKD well, you might never need it. Many people live into their 70s with normal function-because they started early.
Final Thought: Knowledge Is Your Shield
PKD is genetic. You didn’t cause it. But you can control how it affects you.
Get tested if you have family history. Monitor your blood pressure like your life depends on it-because it does. Drink water. Avoid ibuprofen. Say no to smoking. Ask for genetic counseling if you’re thinking about kids.
And if you’re already diagnosed? Don’t wait for symptoms. Don’t wait for pain. Start now. The next decade of your life depends on what you do today.
Is polycystic kidney disease inherited?
Yes, PKD is almost always inherited. Autosomal dominant PKD (ADPKD) is passed down when one parent has the mutated PKD1 or PKD2 gene-each child has a 50% chance of inheriting it. Autosomal recessive PKD (ARPKD) requires both parents to carry the PKHD1 gene mutation. In that case, each child has a 25% chance of developing the disease. About 10% of ADPKD cases happen from new mutations with no family history.
Can you live a normal life with PKD?
Absolutely. Many people with PKD live full, active lives for decades. The key is early detection and strict management: controlling blood pressure, staying hydrated, avoiding kidney-toxic drugs, and getting regular checkups. Some people never need dialysis. Others live into their 70s with good kidney function. It’s not about the diagnosis-it’s about what you do after it.
Does PKD affect other organs besides the kidneys?
Yes. In ADPKD, cysts can form in the liver, pancreas, and even the brain (causing aneurysms). About 80% of people with ADPKD develop liver cysts, though they rarely cause serious problems. ARPKD often leads to severe liver scarring (fibrosis), which can cause portal hypertension and bleeding. Heart valve issues and hernias are also more common in PKD patients.
What’s the difference between PKD1 and PKD2?
PKD1 mutations cause more aggressive disease. People with PKD1 typically reach kidney failure by age 54-58. Those with PKD2 usually don’t need dialysis until age 74-78. PKD1 also causes larger kidneys and more cysts at an earlier age. About 78% of ADPKD cases are PKD1, while 15% are PKD2. The rest are unknown or mixed.
Can PKD be cured?
There is no cure yet. But treatments like tolvaptan can slow progression significantly. A kidney transplant replaces the damaged organ and cures the kidney failure-but it doesn’t remove the gene. The new kidney won’t develop cysts, but the person still carries the mutation. Research into gene therapies and targeted drugs is ongoing, with several promising drugs in late-stage trials.
How often should someone with PKD get checked?
Annual checkups are the minimum: blood pressure, eGFR, and a urine test. If eGFR drops below 60 mL/min, switch to quarterly testing. Imaging (ultrasound or MRI) should be done every 1-3 years to track kidney size and cyst growth. Genetic testing is recommended for family planning, early diagnosis, or if symptoms appear unusually young. Blood pressure should be monitored at home weekly if you’re on medication.
I was diagnosed with ADPKD at 32. No symptoms until then. My dad had it too. Started on losartan, cut out caffeine, drink 3L water a day. BP stays under 120/78. eGFR stable for 4 years. It’s not a death sentence. It’s a lifestyle adjustment. Do the work.
I’ve been reading up on this because my sister just got diagnosed. It’s wild how much the science has changed in the last decade. I used to think it was just about waiting for dialysis, but now I see it’s more about managing the progression. Tolvaptan sounds expensive, but if it buys someone 10 years of kidney function, isn’t that worth fighting insurance for? I’m glad they’re researching alternatives like lixivaptan. The fact that hydration and blood pressure control can slow it down by 14% is huge. I wish more doctors talked about this like a marathon, not a countdown.
The clinical data presented here is methodologically sound. Blood pressure targets below 130/80 are supported by HALT-PKD and AASK trials. The use of ACE inhibitors and ARBs as renoprotective agents is well-documented. However, the cost-effectiveness of tolvaptan remains contentious in real-world settings due to high drug expenditure and hepatic side effects. A structured, longitudinal monitoring protocol with annual eGFR and MRI is optimal for early intervention.
You guys are being played. Tolvaptan was pushed by Big Pharma because they knew people would panic and pay anything. The real issue? The FDA approved it based on surrogate endpoints, not hard outcomes like transplant avoidance. And guess what? The liver toxicity is real. I’ve seen three people end up in the hospital from it. Meanwhile, the real solution-lifestyle changes-is ignored because it doesn’t make money. Wake up.
This whole post is a government psyop. PKD isn’t genetic. It’s caused by chemtrails and fluoridated water. They’re just using this to sell expensive drugs and push mandatory genetic testing so they can track your DNA. I looked up the NIH funding for PKD research-over $200M in 5 years. Who benefits? Pharma. Who pays? You. Don’t get scanned. Don’t take the pill. Drink spring water. Avoid hospitals. Truth is hidden.
The article is technically accurate but disappointingly superficial. It omits critical nuances: the heterogeneity in cyst growth rates even within PKD1 subtypes, the lack of long-term data on tolvaptan beyond 5 years, and the absence of discussion on sex-based differences in progression (females with PKD1 often progress slower due to estrogen’s protective effect). Also, the mention of transplant wait times ignores regional disparities-someone in rural Alabama waits 2x longer than someone in Boston. This is not a public service. It’s a glossy brochure.
I’ve had PKD since birth. My kidneys are 28 pounds. I’ve been on dialysis for 3 years. I’ve had 3 infections from cysts. I’ve had 7 surgeries. My wife left me because I couldn’t work. My kids got tested-both have the gene. My mom died at 52. My dad at 58. I’m 49. I’m not scared of dying. I’m scared of how much this has stolen from me. No one talks about the loneliness. The pain that doesn’t go away with pills. The guilt of knowing you passed this on. This article says ‘knowledge is your shield.’ But what if your shield is rusted? What if you’ve done everything right and still lost everything? I’m not a statistic. I’m not a case study. I’m still here.
America’s healthcare system is broken. You need insurance to get tolvaptan. You need money to get an MRI. You need time off work to get checked. Meanwhile, people in other countries get basic care. This isn’t about PKD. It’s about capitalism turning a genetic disease into a profit engine. Stop pretending this is science. It’s a business model.
Respected colleagues, I am a nephrologist practicing in Mumbai. I have managed over 120 PKD cases in the last decade. The principles outlined are universally applicable. However, in low-resource settings, access to MRI and tolvaptan is negligible. We rely on ultrasound, strict BP control, and hydration. The cost of tolvaptan is equivalent to 18 months of median income here. Yet, patients still thrive with disciplined lifestyle management. The real issue is not the disease-it is inequity in access to care. Knowledge is not enough. Compassion must be scalable.
I just read this article and I have to say the grammar is impeccable. The sentence structure is clear, the terminology is precise, and the use of passive voice in clinical contexts is appropriately deployed. However, there is a minor typographical error: '1.3 mL/min/year' should be '1.3 mL/min/1.73 m²/year' to reflect the standard normalization for GFR. Also, 'eGFR' should be spelled out on first use. These are minor, but they matter in academic contexts.
I’ve seen too many people get diagnosed and just give up. This isn’t a terminal illness. It’s a chronic condition. You don’t stop living because your kidneys are messed up. I’m 44, still hiking, still lifting, still working. I drink water, I avoid ibuprofen, I check my BP every week. I don’t need a miracle drug. I need discipline. And if you’re waiting for someone to hand you a cure, you’re already losing.
I’m not saying this is fake, but why is the NIH funding this? Why aren’t they researching why PKD is more common in white populations? And why is the genetic testing only focused on PKD1/PKD2? What about epigenetics? What about environmental triggers? This feels like a cover-up. They’re not trying to cure it-they’re trying to monetize it. And don’t get me started on how they use ‘family history’ to scare people into testing.
I read this and I just felt… seen. Not just the medical stuff, but the quiet grief. The way you hold your breath when the doctor says ‘the cysts are growing.’ The way you look at your kids and wonder if they’ll ever get to be 60 without dialysis. I didn’t cry when I was diagnosed. I cried when I saw my 8-year-old niece ask if she’d get it too. This isn’t just about kidneys. It’s about legacy. I’m not fighting to live longer. I’m fighting so my kids don’t have to live in fear.
To everyone here: you’re not alone. I’ve been mentoring PKD patients for 12 years. Some are on transplant lists. Some are managing well. Some are scared. All of you are brave. I’ve seen people who started walking 30 minutes a day, stopped soda, and held off dialysis for 15 years. It’s not magic. It’s consistency. You don’t need to be perfect. Just show up. One day at a time. I’m here if you need to talk.
I’m a nurse in Lagos. I’ve seen kids with ARPKD in places with no dialysis machines. No CT scans. No tolvaptan. But I’ve seen mothers carry their kids 10km to a clinic just to get a BP check. They don’t have names for their disease. They just call it ‘the swelling.’ And still-they fight. This article is for the West. But the real heroes? They’re not on Reddit. They’re in villages, holding their children’s hands, whispering ‘it’s okay’ even when they don’t know if it is. We need to remember them too.